🧬 Retour aux sources ! Apprendre les acides aminés non polaires, ce que sont réellement les zwitterions et se plonger dans les mathématiques appliquées – rotation de Rodrigues et potentiel de Lennard-Jones. Construisez lentement vers un phi/psi optimal !
Motivations
Nous avons beaucoup exploré ces derniers temps la simulation de dynamique moléculaire, puis l’amarrage protéine-protéine la dernière fois. Il y a encore tellement de choses à apprendre. J’ai décidé de revenir à l’essentiel, de revisiter nos vieux amis les acides aminés et d’essayer de comprendre les propriétés naturelles derrière chacun et de voir si cela aura plus de sens à l’avenir lorsque nous explorerons davantage. Tout en prenant des notes sur tous les acides aminés, j’essaierai également de comprendre certaines des mathématiques de base derrière les structures. Es-tu prêt !? Mdr, ce n’est pas le cas, mais allons-y quand même ! 🤣
Objectifs :
Acides aminés
Les acides aminés sont les éléments constitutifs des protéines, chacun partageant un squelette commun : un carbone α central lié à un groupe aminé (–NH₂), un groupe carboxyle (–COOH), un atome d’hydrogène et une chaîne latérale variable (groupe R) qui définit l’identité et la chimie de chaque acide aminé.

Acides aminés non polaires
Les acides aminés non polaires ont des chaînes latérales hydrophobes : ils évitent l’eau et ont tendance à se regrouper à l’intérieur des protéines repliées, formant le noyau hydrophobe qui assure la stabilité des protéines. Comprendre la forme et le volume de chacun est directement lié à la manière dont ils se conditionnent, à la manière dont ils limitent la flexibilité du squelette et à la manière dont les substitutions affectent les sites actifs enzymatiques.
library(tibble) library(kableExtra) aa_nonpolar <- tribble( ~aa, ~aa3, ~name, ~functional_group, ~smiles_sidechain, ~charge_ph7, ~mw_da, ~pka, ~md_note, ~main_function, "G", "Gly", "Glycine", "H (none)", "[H]", "Neutral", 75.03, NA_real_, "Minimal VDW radius; unrestricted phi/psi; near-zero excluded volume", "Conformational flexibility; tight turns; active site geometry", "A", "Ala", "Alanine", "Methyl", "C", "Neutral", 89.09, NA_real_, "Low steric perturbation; high alpha-helix propensity in force fields", "Helix former; hydrophobic core; alanine-scanning mutagenesis", "V", "Val", "Valine", "Isopropyl", "CC(C)", "Neutral", 117.15, NA_real_, "Beta-branching restricts psi; favors extended beta-sheet; large gamma-carbons", "Beta-sheet core; hydrophobic packing; sickle-cell HbS Glu6Val", "L", "Leu", "Leucine", "Isobutyl", "CCC(C)C", "Neutral", 131.17, NA_real_, "Flexible chi2; common rotamers at -65/-65 and -65/175; high hydrophobic SASA", "Hydrophobic core; leucine zippers; most abundant non-polar in proteomes", "I", "Ile", "Isoleucine", "sec-Butyl", "CCC(C)", "Neutral", 131.17, NA_real_, "Beta-branching + gamma-branch; most restricted chi1/chi2; large buried SASA", "Hydrophobic core; beta-barrel interiors; transmembrane helices", "P", "Pro", "Proline", "Pyrrolidine ring", "C1CCNC1", "Neutral", 115.13, NA_real_, "Fixed phi ~-60; no backbone NH donor; cis/trans isomerism at Xaa-Pro bond", "Helix breaker; beta-turns; collagen Gly-Pro-X repeats", "F", "Phe", "Phenylalanine", "Benzyl", "Cc1ccccc1", "Neutral", 165.19, NA_real_, "Rigid aromatic ring; pi-pi stacking and cation-pi in MD energy decomposition", "Hydrophobic core; aromatic clusters; ligand binding pockets", "W", "Trp", "Tryptophan", "Indolylmethyl", "Cc1c[nH]c2ccccc12", "Neutral", 204.23, NA_real_, "Indole NH can H-bond; amphipathic at membrane interface; strong 280nm absorbance", "Membrane anchoring; fluorescence probe; ligand binding; rarest standard AA", "M", "Met", "Methionine", "Thioether", "CCSC", "Neutral", 149.20, NA_real_, "Flexible sulfur geometry; oxidizable to sulfoxide in long MD runs; check reactive FF", "Translation initiation; hydrophobic core; redox sensing" ) aa_nonpolar |> dplyr::select(aa:mw_da) |> kbl()
|
aa |
aa3 |
nom |
groupe_fonctionnel |
smiles_sidechain |
charge_ph7 |
mw_da |
|---|---|---|---|---|---|---|
|
G |
Gly |
Glycine |
H (aucun) |
[H] |
Neutre |
75.03 |
|
UN |
Hélas |
Alanine |
Méthyle |
C |
Neutre |
89.09 |
|
V |
Val |
Valine |
Isopropyle |
CC(C) |
Neutre |
117.15 |
|
L |
Leu |
Leucine |
Isobutyle |
CCC(C)C |
Neutre |
131.17 |
|
je |
Avec |
Isoleucine |
sec-butyle |
CCC(C) |
Neutre |
131.17 |
|
P. |
Pro |
Proline |
Anneau pyrrolidine |
C1CCNC1 |
Neutre |
115.13 |
|
F |
Phé |
Phénylalanine |
Benzyle |
Cc1cccc1 |
Neutre |
165.19 |
|
W |
Trp |
Tryptophane |
Indolylméthyle |
Cc1c[nH]c2ccccc12 |
Neutre |
204.23 |
|
M |
Rencontré |
Méthionine |
Thioéther |
CCSC |
Neutre |
149.20 |
aa_nonpolar |> dplyr::select(aa,aa3,md_note,main_function) |> kbl()
|
aa |
aa3 |
md_note |
fonction_main |
|---|---|---|---|
|
G |
Gly |
Rayon VDW minimal ; phi/psi sans restriction ; volume exclu proche de zéro |
Flexibilité conformationnelle ; virages serrés; géométrie du site actif |
|
UN |
Hélas |
Faible perturbation stérique ; forte propension à l’hélice alpha dans les champs de force |
Hélice ancienne ; noyau hydrophobe; mutagenèse par balayage d’alanine |
|
V |
Val |
La branche bêta restreint le psi ; privilégie la feuille bêta étendue ; gros carbones gamma |
Noyau de feuille bêta ; emballage hydrophobe; HbS drépanocytaire Glu6Val |
|
L |
Leu |
Chi2 flexible ; rotamères communs à -65/-65 et -65/175 ; SASA hautement hydrophobe |
Noyau hydrophobe ; fermetures éclair à leucine; non polaire le plus abondant dans les protéomes |
|
je |
Avec |
Ramification bêta + branche gamma ; chi1/chi2 le plus restreint ; grande SASA enterrée |
Noyau hydrophobe ; intérieurs de barils bêta ; hélices transmembranaires |
|
P. |
Pro |
Phy fixe ~-60 ; pas de donneur de NH de base ; Isomérie cis/trans chez Xaa-Pro bond |
Brise-hélice ; tours bêta ; collagène Gly-Pro-X répète |
|
F |
Phé |
Anneau aromatique rigide ; empilement pi-pi et cation-pi dans la décomposition énergétique MD |
Noyau hydrophobe ; grappes aromatiques; poches de liaison de ligand |
|
W |
Trp |
Indole NH peut se lier en H ; amphipathique à l’interface membranaire; forte absorbance de 280 nm |
Ancrage membranaire ; sonde fluorescente ; liaison du ligand ; AA standard le plus rare |
|
M |
Rencontré |
Géométrie flexible du soufre ; oxydable en sulfoxyde lors de longues séries MD ; vérifier le FF réactif |
Initiation à la traduction ; noyau hydrophobe; détection redox |
Claude a généré la plupart des informations ci-dessus. Nous ajouterons des éléments à la section md_note au fur et à mesure que nous rencontrerons certaines choses lors de nos simulations MD.
Qu’est-ce que Zwitterion ?
Un zwitterion est une molécule qui possède à la fois des charges positives et négatives mais qui est globalement électriquement neutre. Dans les acides aminés, le groupe amino (–NH₂) peut accepter qu’un proton devienne chargé positivement (–NH₃⁺), tandis que le groupe carboxyle (–COOH) peut perdre un proton pour devenir chargé négativement (–COO⁻). À un pH physiologique (~ 7,4), la plupart des acides aminés existent sous forme de zwitterions, avec le groupe amino protoné et le groupe carboxyle déprotoné. Cette double charge permet aux acides aminés d’interagir avec les environnements polaires et non polaires, contribuant ainsi à leur solubilité dans l’eau et à leur capacité à former diverses interactions dans les protéines.
Que signifie réellement non polaire ?
Il convient de clarifier ce que « non polaire » fait réellement référence exclusivement à la chaîne latérale (groupe R) – en particulier qu’elle est constituée en grande partie de liaisons carbone et hydrogène sans dipôle net ni groupes ionisables, ce qui la rend hydrophobe et largement indifférente à l’eau. Cela ne dit rien sur le squelette, qui est le même pour tous les acides aminés et porte toujours des liaisons polaires (C=O, N-H). En fait, comme mentionné ci-dessus, tous les acides aminés, y compris les acides non polaires, existent sous forme de zwitterions au pH physiologique – une propriété qui provient entièrement du squelette et non de la chaîne latérale.
Note à moi-même : les squelettes de tous les acides aminés sont des zwitterions ; la chaîne du côté R détermine la polarité et l’hydrophobie. De plus, charge nette neutre == les charges globales sont égales à zéro, cela ne signifie pas que la molécule est non polaire.
Formule de rotation Rodriguez
La formule de rotation de Rodrigues est une méthode permettant de faire pivoter un vecteur 3D dans l’espace autour d’un axe spécifié d’un angle donné. La formule s’exprime ainsi :
\(v_{rotation} = v.\cos(\theta) + \sin(\theta)(k \times v) + (1 - \cos(\theta))(k(k \cdot v))\)
où v est le vecteur d’origine, k est le vecteur unitaire le long de l’axe de rotation, et θ est l’angle de rotation en radians.


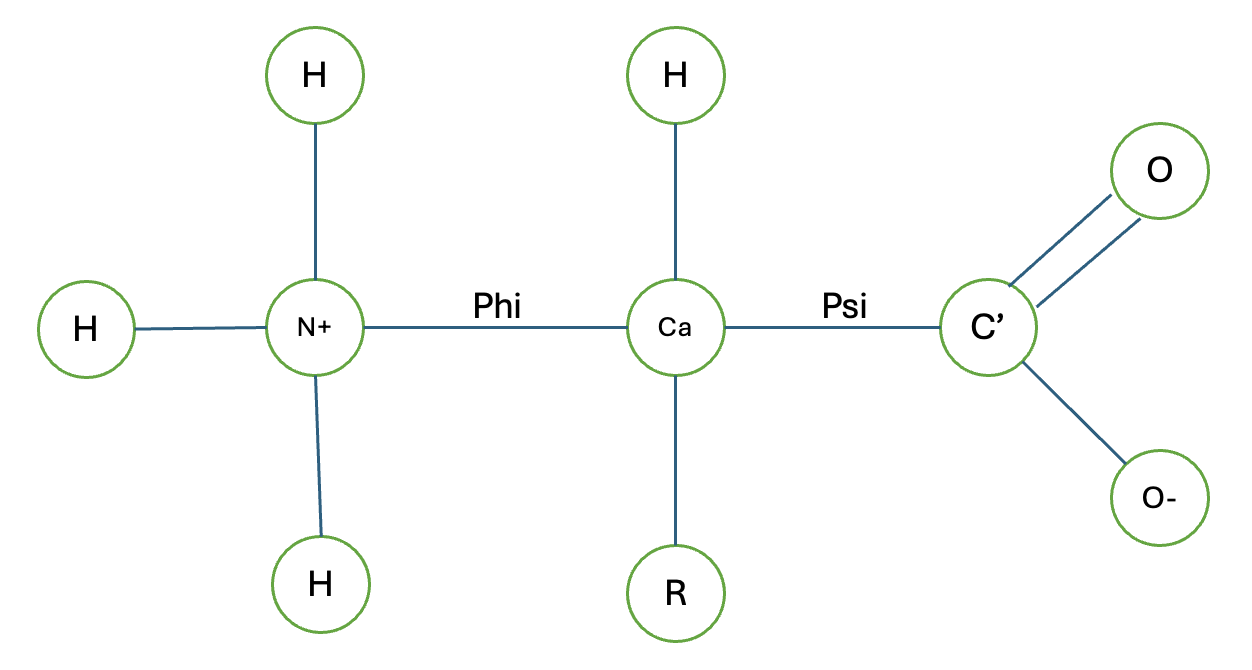
Cette formule est apparemment très populaire en infographie et en robotique, mais je peux voir comment elle peut également être utile en dynamique moléculaire lorsque nous voulons faire tourner une molécule ou une partie de celle-ci autour d’un axe. Surtout quand on veut estimer la conformation de moindre énergie d’une molécule. L’application directe de cette formule dans la séquence d’acides aminés consisterait à réorganiser les atomes en fonction de phi et psi qui sont les angles de rotations autour du N-Cα et Cα-C liaisons du squelette des acides aminés, respectivement. En appliquant la formule de rotation de Rodrigues, nous pouvons calculer les nouvelles positions des atomes dans l’acide aminé après les avoir fait pivoter selon les angles spécifiés, ce qui nous permet d’explorer différentes conformations de la molécule. Comment je me souviens de quel angle est lequel est Nancy Phi (on dirait une émission policière et aussi N->C) et C C Psi (Le tout avec le son S, également Carbon to Carbon). Nous laisserons le calcul manuel jusqu’à la prochaine fois, mais apprenons à faire pivoter une coordonnée basée sur un axe avec Rodriguez !
Ci-dessous, je vais d’abord écrire le code, puis l’expliquer. N’hésitez pas à utiliser votre souris pour survoler l’objet tracé et vérifier les coordonnées.
library(plotly)
library(pracma)
#### Let's start simple
x1 <- c(0,0,0)
x2 <- c(1,1,1)
x3 <- c(1,2,1)
rodrigues <- function(v, k, theta) {
k <- k / sqrt(sum(k^2))
cos(theta)*v + sin(theta)*pracma::cross(k, v) + (1 - cos(theta))*sum(k*v)*k
}
k <- x2 - x1
v <- x3 - x1
result <- rodrigues(v,k,pi/2) #notice this, pi/2 == 90 degrees
pts <- data.frame(
x = c(x1[1],x2[1],x3[1]),
y = c(x1[2],x2[2],x3[2]),
z = c(x1[3],x2[3],x3[3]),
label = c("x1","x2","x3")
)
pts_rs <- data.frame(
x = c(x1[1],x2[1],result[1]),
y = c(x1[2],x2[2],result[2]),
z = c(x1[3],x2[3],result[3]),
label = c("x1","x2","x3_new")
)
plot_ly() |>
add_trace(data=pts, x=~x, y=~y, z=~z,
type="scatter3d", mode="lines+markers+text",
text=~label,
marker=list(size=8, color="blue", opacity=0.5),
line=list(width=4, color="blue", dash="solid")) |>
add_trace(data=pts_rs, x=~x, y=~y, z=~z,
type="scatter3d", mode="lines+markers+text",
text=~label,
marker=list(size=8, color="red", opacity=0.5),
line=list(width=4, color="red", dash="dash"))
Donc, avec ce qui précède, nous voulons commencer avec 3 points, x1, x2, x3. ils représentent tous leurs coordonnées xyz.
Ce qui est drôle, c’est que la coordonnée xyz ici est différente de ce que j’ai appris comme xyz. J’ai toujours pensé que x était horizontal, y était vertical et z était la profondeur. Mais dans ce cas, x est la profondeur, y est horizontal et z est vertical. Je suppose que cela dépend de la façon dont vous le voyez.
Ensuite, nous voulons faire pivoter x3 autour de l’axe défini par x1 et x2 de 90 degrés (pi/2 radians). Le rodrigues la fonction prend le vecteur v (qui est le vecteur de x1 à x3), l’axe k (qui est le vecteur de x1 à x2), et l’angle theta (qui est pi/2). Il renvoie les nouvelles coordonnées de x3 après rotation.
Enfin, nous traçons les points d’origine et le point pivoté en utilisant plotly. Les points d’origine sont en bleu et le point pivoté est en rouge. Vous pouvez survoler les points pour voir leurs coordonnées.
Maintenant, si nous devions manœuvrer le tracé 3D et aligner x1 et x2 en un point, nous pouvons clairement voir qu’il s’est déplacé de 90 degrés dans le sens inverse des aiguilles d’une montre !

Maintenant, il existe une règle plutôt sympa pour savoir où la rotation doit se produire dans le sens inverse des aiguilles d’une montre ou dans le sens des aiguilles d’une montre, c’est en utilisant votre main !!! Tu te souviens de ça au lycée ?

Ce serait vraiment cool de dériver la formule ci-dessus. Il existe de nombreuses vidéos qui font cela. J’essaie encore de le conceptualiser, laissons ça pour un autre blog ! Cela semble intéressant et peut être un bon exercice, surtout lorsque nous nous aventurons dans des espaces 3D.
Énergie potentielle de Lennard-Jones
La formule de l’énergie potentielle de Lennard-Jones est un modèle mathématique utilisé pour décrire l’interaction entre une paire d’atomes ou de molécules neutres. Elle est donnée par l’équation :
\(V(r) = 4\epsilon \left[ \left( \frac{\sigma}{r} \right)^{12} - \left( \frac{\sigma}{r} \right)^6 \right]\)
Où:
\(V(r)\)est l’énergie potentielle en fonction de la distance\(r\)entre les deux particules.\(\epsilon\)est la profondeur du puits de potentiel, représentant la force de l’interaction attractive.\(\sigma\)est la distance finie à laquelle le potentiel inter-particules est nul, représentant le diamètre effectif des particules.- Le terme
\(\left( \frac{\sigma}{r} \right)^{12}\)représente la partie répulsive du potentiel, qui domine sur de courtes distances en raison du principe d’exclusion de Pauli. - Le terme
\(\left( \frac{\sigma}{r} \right)^6\)représente la partie attractive du potentiel, qui domine à de plus longues distances en raison des forces de Van der Waals.
Le potentiel de Lennard-Jones est largement utilisé dans les simulations de dynamique moléculaire pour modéliser les interactions entre atomes ou molécules non liés, notamment dans le contexte des forces de van der Waals. Cela aide à prédire le comportement des particules dans un système, comme leurs positions d’équilibre et le paysage énergétique des interactions moléculaires.
Wow, il y a un tas de termes et de mots ci-dessus ! J’ai le vertige rien que de savoir ce qui se passe. Allons-y. Pour utiliser la formule ci-dessus, nous devons avoir une certaine compréhension des paramètres epsilon et sigma. Ces paramètres sont généralement dérivés de données expérimentales ou de calculs de mécanique quantique et sont spécifiques aux types d’atomes ou de molécules impliqués dans l’interaction. Où trouver ces paramètres ? Et voilà – openbabel :: gaff.dat
Quand tu as ouvert gaff.dat il y a plein de chiffres ! Trouvons les chiffres qui sont significatifs pour nous. Tous les éléments ci-dessous sont séparés par de nouvelles lignes au fur et à mesure que vous faites défiler vers le bas.
Étirement des liaisons
Les noms de colonnes doivent être : type mass(g/mol) polarizability(ų) source

Angle de liaison
Les noms de colonnes doivent être : types K r0 source count rmsd

Dièdre approprié
Les noms de colonnes doivent être : types div barrier phase periodicity

Non collé
Les noms de colonnes doivent être : type R*(Å) ε(kcal/mol)

Nous nous intéressons uniquement à non-bonded section où R* est notre sigma = R* × 2 / 2^(1/6) et ε est notre epsilon. Avec les paramètres ci-dessus, nous pouvons alors calculer l’énergie potentielle de Lennard-Jones entre deux atomes quelconques d’une molécule. Faisons un calcul simple pour l’éthanol.
Calculer LJ
library(tidyverse)
library(igraph)
# Coordinates (x, y, z) in Angstroms, according to pubchem ethanol molecule
coords <- rbind(
O = c( -1.1712, 0.2997, 0.0000),
C2 = c( -0.0463, -0.5665, 0.0000),
C1 = c( 1.2175, 0.2668, 0.0000),
H4 = c( -0.0958, -1.2120, 0.8819),
H5 = c( -0.0952, -1.1938, -0.8946),
H1 = c( 2.1050, -0.3720, -0.0177),
H2 = c( 1.2426, 0.9307, -0.8704),
H3 = c( 1.2616, 0.9052, 0.8886),
H6 = c( -1.1291, 0.8364, 0.8099)
)
# AMBER GAFF parameters (sigma Å, epsilon kcal/mol)
Rstar_to_sigma <- function(Rstar) 2 * Rstar / 2^(1/6)
sigma <- c(
C1=Rstar_to_sigma(1.9080), C2=Rstar_to_sigma(1.9080),
O=Rstar_to_sigma(1.7210),
H1=Rstar_to_sigma(1.4870), H2=Rstar_to_sigma(1.4870),
H3=Rstar_to_sigma(1.4870), H4=Rstar_to_sigma(1.4870),
H5=Rstar_to_sigma(1.4870), H6=0.0000
)
epsilon <- c(
C1=0.1094, C2=0.1094, O=0.2104,
H1=0.0157, H2=0.0157, H3=0.0157,
H4=0.0157, H5=0.0157, H6=0.0000
)
# Bonds
bonds <- tribble(
~from, ~to,
"C1", "C2",
"C2", "O",
"O", "H6",
"C1", "H1",
"C1", "H2",
"C1", "H3",
"C2", "H4",
"C2", "H5"
)
# count bonds between two atoms
g <- graph_from_data_frame(bonds, directed = FALSE)
g_dist <- distances(g)
# LJ function
lj <- function(r, eps, sig) 4 * eps * ((sig/r)^12 - (sig/r)^6)
# Loop all pairs
atoms <- rownames(coords)
pairs <- combn(atoms, 2, simplify=FALSE) # combn so we don't repeat
total_V <- vector(mode = "numeric", length = length(pairs))
for (i in 1:length(pairs)) {
# each pair
p <- pairs[[i]]
from <- p[1]
to <- p[2]
num_bond <- g_dist[from, to]
if (num_bond <= 2) next
# params needed for LJ
r <- sqrt(sum((coords[from,] - coords[to,])^2))
sig <- (sigma[from] + sigma[to]) / 2
eps <- sqrt(epsilon[from] * epsilon[to])
# scale if num bond is 3 (4 atoms)
scale <- if (num_bond == 3) 0.5 else 1.0
# LJ calc
V <- scale * lj(r, eps, sig)
cat(from, "-", to, " num of bonds=", num_bond, " r=", r, " V=", V, "\n")
total_V[i] <- V
}
## O - H1 num of bonds= 3 r= 3.344395 V= -0.02733269
## O - H2 num of bonds= 3 r= 2.642383 V= 0.110613
## O - H3 num of bonds= 3 r= 2.659841 V= 0.09535471
## C1 - H6 num of bonds= 3 r= 2.546942 V= 0
## H4 - H1 num of bonds= 3 r= 2.521587 V= 0.01461147
## H4 - H2 num of bonds= 3 r= 3.074579 V= -0.007593085
## H4 - H3 num of bonds= 3 r= 2.514978 V= 0.01576022
## H4 - H6 num of bonds= 3 r= 2.295394 V= 0
## H5 - H1 num of bonds= 3 r= 2.507028 V= 0.01720963
## H5 - H2 num of bonds= 3 r= 2.510736 V= 0.01652426
## H5 - H3 num of bonds= 3 r= 3.070262 V= -0.007612401
## H5 - H6 num of bonds= 3 r= 2.845344 V= 0
## H1 - H6 num of bonds= 4 r= 3.550289 V= 0
## H2 - H6 num of bonds= 4 r= 2.908137 V= 0
## H3 - H6 num of bonds= 4 r= 2.392984 V= 0
cat("\nTotal V_LJ:", sum(total_V), "kcal/mol\n")
##
## Total V_LJ: 0.2275351 kcal/mol
Très bien, ce qui précède, nous essayions essentiellement de calculer tous les atomes par paires qui comportent plus de 3 liaisons (s’il s’agit exactement de 3 liaisons, nous le réduisons de moitié). Très bien, maintenant que nous savons comment faire cela sur des molécules simples, la prochaine fois, nous pourrons utiliser cela pour minimiser car nous recherchons un phi et un psi optimaux !
Possibilités d’amélioration
- Dériver la formule de rotation de Rodriguez
- Mettez en action la formule de rotation de Rodriguez et le calcul LJ pour trouver le phi et le psi optimaux de la séquence d’acides aminés d’une protéine !
- il faut également inclure les interactions entre structures secondaires et tertiaires
Leçons apprises
- rafraîchi sur la rotation et les vecteurs
- appris la formule de rotation de Rodriguez
- appris la formule LJ
- appris ce que contient réellement gaff.dat.
- J’ai découvert le phi et le psi et ce qu’ils signifient réellement.
- connaissance de la charge nette == charge totale ; polaire vs non polaire dépend de la chaîne R.
Si vous aimez cet article :
En rapport
PakarPBN
A Private Blog Network (PBN) is a collection of websites that are controlled by a single individual or organization and used primarily to build backlinks to a “money site” in order to influence its ranking in search engines such as Google. The core idea behind a PBN is based on the importance of backlinks in Google’s ranking algorithm. Since Google views backlinks as signals of authority and trust, some website owners attempt to artificially create these signals through a controlled network of sites.
In a typical PBN setup, the owner acquires expired or aged domains that already have existing authority, backlinks, and history. These domains are rebuilt with new content and hosted separately, often using different IP addresses, hosting providers, themes, and ownership details to make them appear unrelated. Within the content published on these sites, links are strategically placed that point to the main website the owner wants to rank higher. By doing this, the owner attempts to pass link equity (also known as “link juice”) from the PBN sites to the target website.
The purpose of a PBN is to give the impression that the target website is naturally earning links from multiple independent sources. If done effectively, this can temporarily improve keyword rankings, increase organic visibility, and drive more traffic from search results.

